


Research effort in the last two decades revealed transcriptional activity within non-coding and coding regions of the entire human genome. This led to the identification of non-coding transcripts that are mainly located in the cell nucleus and expressed at lower levels than coding-RNAs [1-4]. The lack of functional annotations made the classification of the different non-coding RNA populations rather difficult. Indeed, an arbitrary cut-off based on the length of non-coding RNAs distinguished long non-coding RNAs (lncRNAs, 100-200nt) from microRNAs (miRNAs), small interfering RNAs (siRNAs), and Piwi-interacting RNAs (piRNAs) that span 21-35 nucleotides respectively [5]. To date, microRNAs are those non-coding RNAs that were mostly studied and closely linked to human cancers [6-9]. MicroRNAs are evolutionarily conserved small non-coding RNAs that regulate gene expression at the posttranscriptional level. This occurs through imperfect complementarity to the 3′untranslated region (3′UTR) of target mRNAs which results in mRNAs translational inhibition and/or degradation and leads ultimately to a reduction in protein expression level. MicroRNAs are predicted to target over 50% of all human protein-coding genes and each gene could be controlled by different microRNAs [10]. Thus, many if not all, cellular functions can be putatively subjected to microRNA control. MicroRNAs were originally identified as regulators of developmental processes including developmental timing and cell fate transitions [11, 12]. Croce’s group originally reported the involvement of microRNAs in chronic lymphocytic leukemia (CLL) [13]. Many miRNAs map to specific regions of the human genome frequently deleted or amplified in human cancers [14-16]. Growing evidence has shown that miRNAs might be differentially expressed in cancer cells, in which they form unique expression patterns or signatures [7]. Altered expression of miRNAs in cancers can occur through epigenetic changes, including aberrant DNA methylation and histone modifications, aberrant transcriptional regulation and genetic alterations [17, 18]. These alterations can affect the production of the primary RNAs, their processing to the mature miRNA forms, and/or interactions with mRNA targets. Mutations in the genes encoding for proteins involved in the processing and maturation of miRNAs such as TARBP2, DICER1 and XPO5 have been found to lead to overall reductions in miRNA expression [19-21]. miRNAs can either act as tumor suppressor genes or oncogenes [22]. Increasing evidence indicate that the expression of miRNAs is mainly downregulated in tumor tissues, as compared to corresponding healthy tissues, which might suggest that miRNAs are primarily tumor suppressor genes [23, 24]. Extensive down-regulation of multiple miRNAs followed rapidly EGF stimulation of breast cancer cells [23]; this was paired by the induction of target mRNAs with oncogenic activities [23]. Several well-characterized oncogenic miRNAs were in tumors. The expression of miRNA 21-5p is very frequently up-regulated in diverse tumoral tissues when compared to both matched or unmatched non-tumoral ones [25, 26]. miRNA 21-5p overexpression promotes tumor growth and invasion [27-30]. Ectopic expression of miR-7 promotes cell growth and tumor formation in lung cancer cells [31]. EGFR activation induces miR-7 expression through a RAS-MYC pathway. Indeed MYC binds to and activates the miR-7 promoter. Aberrant transcriptional regulation of miRNA-10b5p by Twist promotes breast cancer metastatization [32]. Exposure to both metabolic cancer risk factors and to carcinogenic substances such as asbestos, formaldehyde and cigarette smoke in lung and hepatic tissues alters miRNA expression[33-37]. This might unveil miRNAs as both predictors and players of cancer development.
P53 gene is the most frequent target for genetic alterations in human cancers [38]. Indeed, more than half of human cancers carry p53 mutations. Most of these mutations are missense and reside in the core domain of p53 protein [39]. They can be roughly divided in two main classes: (a) DNA contact defective mutants whose residue subjected to mutation is located in the region of the protein that binds to DNA; (b) Structural defective mutants whose mutation impinges on a residue critical for the entire folding of the protein [40]. There are two main features that distinguish wt-p53 and mutant p53 proteins. Mutant p53 proteins are unable to bind to wt-p53 DNA binding consensus and consequently are unable to promote transcriptional dependent wt-p53 tumor suppressor activities. Mutant p53 are rather stable proteins as their half-life is extremely prolonged when compared to that of wt-p53 protein. While in the past decades mutant p53 proteins were mainly considered as loss of function gene products with no specific activities, increasing evidence have established that these proteins gain additional functions through which strongly contribute the transformed phenotype of a given tumor cell [41-43]. Since mutant p53 proteins are abundantly present in many human cancers, the rationale to envisage them as an important target for novel cancer therapeutic intervention is very strong and consequently is arousing a remarkable interest. This also led to extraordinary experimental effort to decipher the molecular mechanisms underlying the oncogenic activities of mutant p53 proteins (Figure 1). Mutant p53 proteins can aberrantly modulate the expression of genes acting as oncogenic transcription factors. Di Agostino et al., originally reported that mutant p53 physically interacted with the transcription factor NF-Y [44]. This led to the transcriptional upregulation of the expression of cell cycle regulated genes such as cyclin B, cdk1 and cdc25. Notably, mutant p53 proteins were recruited onto the DNA binding consensus for the transcription factor NF-Y, thus implying its direct involvement on transcriptional machinery that leads to the aberrant regulation of NF-Y target genes. Since these observations, other transcriptional crosstalks involving other transcription factors such as VDR, SP1, E2F1, ETS1, NFKb, were reported [45-49]. These findings implicate that gain of function mutant p53 proteins can broadly modulate gene expression as they can parasite the activity of the interacting transcription factors. Mutant p53 proteins could putatively modulate the expression of most, if not all, gene targets of the parasite transcription factors. There is still very scarce evidence on which is the role of mutant p53 proteins in the context of these oncogenic transcriptional competent complexes. It was originally reported that mutant p53 protein in complex with the transcription factor NFY favored the recruitment of the acetylase p300; thereby potentiating the activity of the interacting transcription factor [44]. Gain of function mutant p53 proteins disabled the transcriptional repressor complex p73/NFY assembled on PDGFr promoter. This led to pancreatic cancer metastasis [50]. In advanced cancer lesions TFGβ ligands act as pro-metastatic factors. Adorno et al., reported that oncogenic Ras and mutant p53 favored the assembling of a protein complex mutant p53/p63 via Smad proteins [51]. This antagonized p63 tumor suppressor activities by impairing its transcriptional function [51]. Mutant p53 and p300 were co-recruited at the promoter of REGg, a proteasome activator, which is overexpressed in tumors and promotes degradation of p53, p21 and p16 [52]. In that context, mutant p53 also prevented the formation of Smad3/N-CoR complex on the REGg promoter and thereby enhancing the activity of the REGg-proteasome within gain of function of mutant p53 proteins. It was recently reported that mutant p53 protein transcriptionally activates SREBP gene and through it aberrantly regulates the mevalonate pathway [53]. Notably, increased levels of mevalonate by mutant p53 proteins promoted nuclear localization and oncogenic activation of two nodal transducers of HIPPO tumor suppressor pathway, named YAP and TAZ [54]. Additional partners can be present in the mutant p53 transcriptional competent complexes and potentiate its activity. The prolyl-isomerase PIN1 enhanced mutant p53 transcriptional activity and conferred gene target specificity in breast cancer cells [55].
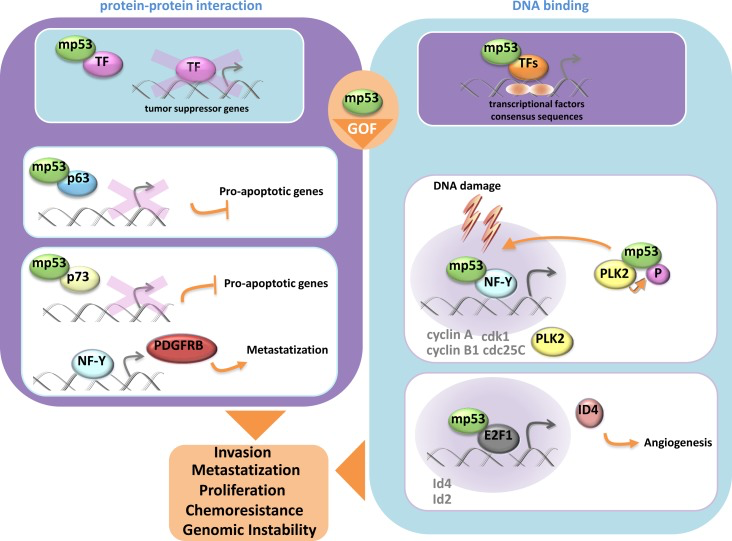
Figure 1. Mutant p53protein gain of function activity
Donzelli et al., originally reported that gain of function mutant p53 proteins can also modulate the expression of microRNAs [56]. Mutant p53 proteins were found on the promoter of ARPP21, the host gene of the oncogenic miR-128b. Aberrant expression of miR-128b contributed to increase chemoresistance of lung cancer cells by targeting the transcriptional repressor E2F5 [56]. This led to increased cytoplasmic accumulation of p21 that in turn reduced the apoptotic rate of lung cancer cells to commonly used anticancer treatments. Masciarelli et al demonstrated that gain of function mutant p53 proteins transcriptionally down-regulated the expression of miR-223 [57]. This resulted in the augmented expression of its mRNA target stathmin-1, an oncoprotein known to confer increased chemoresistance and associated with poor clinical prognosis [57]. Interestingly, a transcriptional cross-talk with mutant p53 and the transcriptional repressor ZEB1, occurring on the regulatory regions of miR-223, was also documented [57]. A direct involvement of gain of function mutant p53 proteins in the processing of microRNAs has been recently evidenced. Mutant p53 proteins downregulated Dicer expression thereby conferring to cancer cells a more invasive and metastatic phenotype as for Dicer depletion [58]. It was recently reported that mutant could promote epithelial-mesenchymal transition (EMT) and tumor metastasis by transcriptionally inhibiting miR-130 that has among its mRNA target the transcriptional repressor ZEB1 [59]. Reduced expression of miR-130 released ZEB1-mediated EMT [59]. Mutant p53-induced up-regulation of miR-155 favored invasion of breast cancer cells [60]. MicroRNA-let7i was shown to be transcriptionally downregulated by diverse mutant p53 proteins. This led to increased invasion and migration of breast cancer cells [61]. MicroRNA-27 was reported to be transcriptionally downregulated by mutant p53 protein [62]. This released aberrant expression of its mRNA target, EGFR, which consequently instigated an increased and prolonged signaling promoting tumor development [62]. The tumor spectrum on both p53 null mice and p53 His172 knock-in mice is similar, but the tumors developed by the latter gain the ability to metastatize. The transcriptional activity of mutant p53 proteins as the ability to modulate both coding and non-coding RNAs appears to play a critical role in mutant p53-mediated tumorigenesis (figure 2). Many studies have reported that patients with tumors carrying p53 mutations have worse prognosis, shorter survival and poorer response to conventional anticancer treatments that those bearing wt-p53 protein [43, 63]. P53 mutations frequently associated with aberrant, either up- or down-regulated, expression of their transcriptional target genes [64]. Ganci et al., recently reported that TP53 mutations associated with a shorter recurrence-free survival of head and neck patients [65]. The expression pattern of 49 miRs associated with TP53 status. In particular, within the 49 miRs, a group of 12 miRs correlated with recurrence free-survival and a group of 4 miRs with cancer-specific survival [65]. Altogether these findings indicate that specific microRNA expression associated with TP53 missense mutations and with reduced survival after surgical treatment of patients with head and neck squamous cell carcimonas.
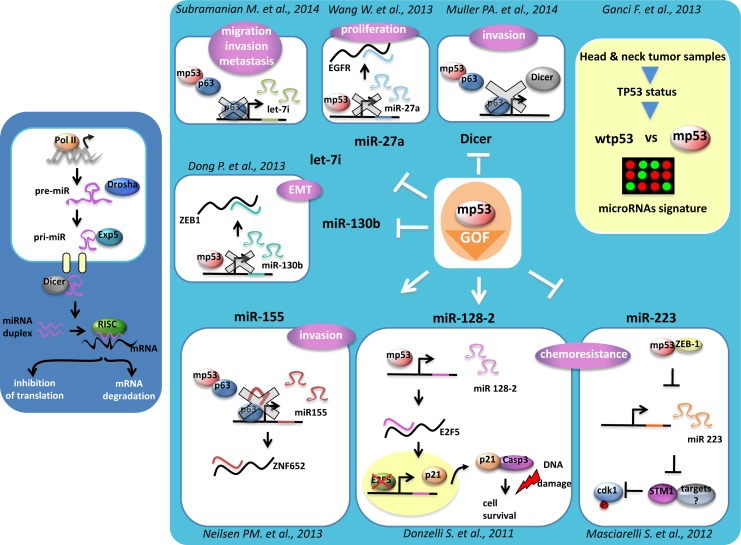
Figure 2. MicroRNAs regulation by mutant p53 protein
Most of the existing evidence for p53 mutations is related to missense mutations, but other type of mutations, such as non-sense and frameshifts are also present in human cancers. The understanding of their contribution to cancer phenotype will provide a comprehensive view of the role of TP53 alterations in human cancers. This also needs to be referred to the presence of the p53 family members p73 and p63 and its derived isoforms that compose a network of more than 20 polypeptides floating in cancer cells. It was originally shown that mutant p53 proteins can physically associate with either p73 or p63 and through it impaired p73- or p63 transcriptional activation and consequently their antitumoral activities [66-69]. Small interfering peptides disassembling the oncogenic protein complex mutp53/ p73 restored p73 oncosuppressor activities [70, 71]. This leads to propose that protein/protein interactions are critical for the oncogenic activities of mutant p53 proteins either to parasite or to inhibit the activity of transcription factors. In such a functional context, the core domain of mutant p53, where most of mutations reside, plays an important role as an interacting platform. Since the mutation of a single p53 residue impacts differently on the overall structure of the protein it can be hypothesized that diverse interacting platforms as for the diverse p53 mutations are present in human cancers. Little is known about the determinants that dictate which are the preferential interacting proteins of a given mutant p53 platform. This may impart specificity in the selection of which transcriptional target will be modulated by gain of function mutant p53 protein. For instance, the inability of mutant p53 proteins to transcriptionally active Mdm2 gene 431 has a profound impact on their half-life. Unlike wt-p53 protein that is extensively subjected to E3 ligase activity of MDM2, mutant p53 proteins evade such a tight control and consequently become rather stable and abundant in cancer cells [72, 73]. This might also occur for other negative regulators that cannot target mutant p53 proteins either because their binding affinity is lower than that to wt-p53 protein or mutant p53 is hidden in “protective” complexes with chaperons thereby making it unavailable to the degradation machinery [74]. It was also reported that mutant p53 proteins are unstable in normal tissues; thereby implying a critical role of the transformed cell context in the stabilization and oncogenic activities of mutant p53 proteins. The contribution of the transformed cell context to the oncogenic activity of mutant p53 proteins could also depend from the organ site of the tumor and from the specificity of the additional genetic alterations that occur in a given tumors. The full comprehension of the molecular events underlying gain of function of mutant p53 proteins is essential for improving: (a) the ongoing therapeutic approaches tackling mutant p53 gain of function; (b) the design of novel mutant p53 personalized cancer therapeutic approaches.
References
- 1. Dogini DB, Pascoal VD, Avansini SH, Vieira AS, Pereira TC, Lopes-Cendes I. The new world of RNAs.Genet Mol Biol.2014;37:285-293. [PubMed] https://doi.org/10.1590/s1415-47572014000200014
- 2. Paschoal AR, Maracaja-Coutinho V, Setubal JC, Simoes ZL, Verjovski-Almeida S, Durham AM. Non-coding transcription characterization and annotation: a guide and web resource for non-coding RNA databases.RNA Biol.2012;9:274-282. [PubMed] https://doi.org/10.4161/rna.19352
- 3. Gezer U, Ozgur E, Cetinkaya M, Isin M, Dalay N. Long non-coding RNAs with low expression levels in cells are enriched in secreted exosomes.Cell Biol Int.2014; https://doi.org/10.1002/cbin.10301 [PubMed]
- 4. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs.Cell.2009;136:629-641. [PubMed] https://doi.org/10.1016/j.cell.2009.02.006
- 5. Hirose T, Mishima Y, Tomari Y. Elements and machinery of non-coding RNAs: toward their taxonomy.EMBO Rep.2014;15:489-507. [PubMed] https://doi.org/10.1002/embr.201338390
- 6. Nana-Sinkam SP and Croce CM. Clinical applications for microRNAs in cancer.Clin Pharmacol Ther.2013;93:98-104. [PubMed] https://doi.org/10.1038/clpt.2012.192
- 7. Calin GA and Croce CM. MicroRNA signatures in human cancers.Nat Rev Cancer.2006;6:857-866. [PubMed] https://doi.org/10.1038/nrc1997
- 8. Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer.Annu Rev Pathol.2014;9:287-314. [PubMed] https://doi.org/10.1146/annurev-pathol-012513-104715
- 9. Di Leva G and Croce CM. miRNA profiling of cancer.Curr Opin Genet Dev.2013;23:3-11. [PubMed] https://doi.org/10.1016/j.gde.2013.01.004
- 10. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs.Genome Res.2009;19:92-105. [PubMed] https://doi.org/10.1101/gr.082701.108
- 11. Carrington JC and Ambros V. Role of microRNAs in plant and animal development.Science.2003;301:336-338. [PubMed] https://doi.org/10.1126/science.1085242
- 12. Kloosterman WP and Plasterk RH. The diverse functions of microRNAs in animal development and disease.Dev Cell.2006;11:441-450. [PubMed] https://doi.org/10.1016/j.devcel.2006.09.009
- 13. Calin GA and Croce CM. Genomics of chronic lymphocytic leukemia microRNAs as new players with clinical significance.Semin Oncol.2006;33:167-173. [PubMed] https://doi.org/10.1053/j.seminoncol.2006.01.010
- 14. Croce CM. Causes and consequences of microRNA dysregulation in cancer.Nat Rev Genet.2009;10:704-714. [PubMed] https://doi.org/10.1038/nrg2634
- 15. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers.Proc Natl Acad Sci U S A.2004;101:2999-3004. [PubMed] https://doi.org/10.1073/pnas.0307323101
- 16. Sacconi A, Biagioni F, Canu V, Mori F, Di Benedetto A, Lorenzon L, Ercolani C, Di Agostino S, Cambria AM, Germoni S, et al. miR-204 targets Bcl-2 expression and enhances responsiveness of gastric cancer.Cell Death Dis.2012;3:e423 [PubMed] https://doi.org/10.1038/cddis.2012.160
- 17. Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, et al. microRNAs exhibit high frequency genomic alterations in human cancer.Proc Natl Acad Sci U S A.2006;103:9136-9141. [PubMed] https://doi.org/10.1073/pnas.0508889103
- 18. Wong KY, Yu L, Chim CS. DNA methylation of tumor suppressor miRNA genes: a lesson from the miR-34 family.Epigenomics.2011;3:83-92. [PubMed] https://doi.org/10.2217/epi.10.74
- 19. Davalos V and Esteller M. Rolling the dice to discover the role of DICER in tumorigenesis.Cancer Cell.2012;21:717-719. [PubMed] https://doi.org/10.1016/j.ccr.2012.05.030
- 20. Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function.Nat Genet.2009;41:365-370. [PubMed] https://doi.org/10.1038/ng.317.
- 21. Melo SA, Moutinho C, Ropero S, Calin GA, Rossi S, Spizzo R, Fernandez AF, Davalos V, Villanueva A, Montoya G, et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells.Cancer Cell.2010;18:303-315. [PubMed] https://doi.org/10.1016/j.ccr.2010.09.007
- 22. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors.Dev Biol.2007;302:1-12. [PubMed] https://doi.org/10.1016/j.ydbio.2006.08.028
- 23. Avraham R, Sas-Chen A, Manor O, Steinfeld I, Shalgi R, Tarcic G, Bossel N, Zeisel A, Amit I, Zwang Y, et al. EGF decreases the abundance of microRNAs that restrain oncogenic transcription factors.Sci Signal.2010;3:ra43 [PubMed] https://doi.org/10.1126/scisignal.2000876
- 24. Lopez-Serra P and Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer.Oncogene.2012;31:1609-1622. [PubMed] https://doi.org/10.1038/onc.2011.354
- 25. Selcuklu SD, Donoghue MT, Spillane C. miR-21 as a key regulator of oncogenic processes.Biochem Soc Trans.2009;37:918-925. [PubMed] https://doi.org/10.1042/BST0370918
- 26. Buscaglia LE and Li Y. Apoptosis and the target genes of microRNA-21.Chin J Cancer.2011;30:371-380. [PubMed] https://doi.org/10.5732/cjc.011.10132
- 27. Li L, Zhou L, Li Y, Lin S, Tomuleasa C. MicroRNA-21 stimulates gastric cancer growth and invasion by inhibiting the tumor suppressor effects of programmed cell death protein 4 and phosphatase and tensin homolog.J BUON.2014;19:228-236. [PubMed]
- 28. Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC).Clin Chim Acta.2010;411:846-852. [PubMed] https://doi.org/10.1016/j.cca.2010.02.074
- 29. Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis.Cell Res.2008;18:350-359. [PubMed] https://doi.org/10.1038/cr.2008.24
- 30. Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21- mediated tumor growth.Oncogene.2007;26:2799-2803. [PubMed] https://doi.org/10.1038/sj.onc.1210083
- 31. Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, Lin SC, Chang YC, Lin SY, Chen SJ, et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF.Cancer Res.2010;70:8822-8831. [PubMed] https://doi.org/10.1158/0008-5472.CAN-10-0638
- 32. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer.Nature.2007;449:682-688. [PubMed] https://doi.org/10.1038/nature06174
- 33. Rotllan N and Fernandez-Hernando C. MicroRNA Regulation of Cholesterol Metabolism.Cholesterol.2012;2012:847849 [PubMed] https://doi.org/10.1155/2012/847849
- 34. Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling.Proc Natl Acad Sci U S A.2011;108:9232-9237. [PubMed] https://doi.org/10.1073/pnas.1102281108
- 35. Rager JE, Smeester L, Jaspers I, Sexton KG, Fry RC. Epigenetic changes induced by air toxics: formaldehyde exposure alters miRNA expression profiles in human lung cells.Environ Health Perspect.2011;119:494-500. [PubMed] https://doi.org/10.1289/ehp.1002614
- 36. Elamin BK, Callegari E, Gramantieri L, Sabbioni S, Negrini M. MicroRNA response to environmental mutagens in liver.Mutat Res.2011;717:67-76. [PubMed] https://doi.org/10.1016/j.mrfmmm.2011.03.015
- 37. Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM, De Flora S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke.FASEB J.2009;23:806-812. [PubMed] https://doi.org/10.1096/fj.08-121384
- 38. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers.Science.1991;253:49-53. [PubMed] https://doi.org/10.1126/science.1905840
- 39. Hainaut P and Hollstein M. p53 and human cancer: the first ten thousand mutations.Adv Cancer Res.2000;77:81-137. [PubMed] https://doi.org/10.1016/s0065-230x(08)60785-x
- 40. Bullock AN and Fersht AR. Rescuing the function of mutant p53.Nat Rev Cancer.2001;1:68-76. [PubMed] https://doi.org/10.1038/35094077
- 41. Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ. Gain of function mutations in p53.Nat Genet.1993;4:42-46. [PubMed] https://doi.org/10.1038/ng0593-42
- 42. Oren M and Rotter V. Mutant p53 gain-of-function in cancer.Cold Spring Harb Perspect Biol.2010;2:a001107 [PubMed] https://doi.org/10.1101/cshperspect.a001107
- 43. Sigal A and Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome.Cancer Res.2000;60:6788-6793. [PubMed]
- 44. Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation.Cancer Cell.2006;10:191-202. [PubMed] https://doi.org/10.1016/j.ccr.2006.08.013
- 45. Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, Shiff I, Kogan I, Shay M, Kalo E, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53.Cancer Cell.2010;17:273-285. [PubMed] https://doi.org/10.1016/j.ccr.2009.11.025
- 46. Chicas A, Molina P, Bargonetti J. Mutant p53 forms a complex with Sp1 on HIV-LTR, DNA.Biochem Biophys Res Commun.2000;279:383-390. [PubMed] https://doi.org/10.1006/bbrc.2000.3965
- 47. Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, Terrenato I, Mottolese M, Muti P, Domany E, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis.Nat Struct Mol Biol.2009;16:1086-1093. [PubMed] https://doi.org/10.1038/nsmb.1669
- 48. Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1.J Biol Chem.2001;276:39359-39367. [PubMed] https://doi.org/10.1074/jbc.M103429200
- 49. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld N, et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer.Cancer Cell.2013;23:634-646. [PubMed] https://doi.org/10.1016/j.ccr.2013.03.022
- 50. Weissmueller S, Manchado E, Saborowski M, Morris JPt Wagenblast, E Davis, CA Moon, SH Pfister, NT Tschaharganeh, DF Kitzing, T et al, et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor beta Signaling.Cell.2014;157:382-394. [PubMed] https://doi.org/10.1016/j.cell.2014.01.066
- 51. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis.Cell.2009;137:87-98. [PubMed] https://doi.org/10.1016/j.cell.2009.01.039
- 52. Ali A, Wang Z, Fu J, Ji L, Liu J, Li L, Wang H, Chen J, Caulin C, Myers JN, et al. Differential regulation of the REGgamma-proteasome pathway by p53/TGF-beta signalling and mutant p53 in cancer cells.Nat Commun.2013;4:2667 [PubMed] https://doi.org/10.1038/ncomms3667
- 53. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway.Cell.2012;148:244-258. [PubMed] https://doi.org/10.1016/j.cell.2011.12.017
- 54. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway.Nat Cell Biol.2014;16:357-366. [PubMed] https://doi.org/10.1038/ncb2936
- 55. Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer.Cancer Cell.2011;20:79-91. [PubMed] https://doi.org/10.1016/j.ccr.2011.06.004
- 56. Donzelli S, Fontemaggi G, Fazi F, Di Agostino S, Padula F, Biagioni F, Muti P, Strano S, Blandino G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function.Cell Death Differ.2012;19:1038-1048. [PubMed] https://doi.org/10.1038/cdd.2011.190
- 57. Masciarelli S, Fontemaggi G, Di Agostino S, Donzelli S, Carcarino E, Strano S, Blandino G. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells.Oncogene.2014;33:1601-1608. [PubMed] https://doi.org/10.1038/onc.2013.106
- 58. Muller PA, Trinidad AG, Caswell PT, Norman JC, Vousden KH. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype.J Biol Chem.2014;289:122-132. [PubMed] https://doi.org/10.1074/jbc.M113.502138
- 59. Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, Sudo S, Ju J, Sakuragi N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis.Oncogene.2013;32:3286-3295. [PubMed] https://doi.org/10.1038/onc.2012.334
- 60. Neilsen PM, Noll JE, Mattiske S, Bracken CP, Gregory PA, Schulz RB, Lim SP, Kumar R, Suetani RJ, Goodall GJ, Callen DF. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155.Oncogene.2013;32:2992-3000. [PubMed] https://doi.org/10.1038/onc.2012.305
- 61. Subramanian M, Francis P, Bilke S, Li XL, Hara T, Lu X, Jones MF, Walker RL, Zhu Y, Pineda M, et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis.Oncogene.2014; https://doi.org/10.1038/onc.2014.46 [PubMed]
- 62. Wang W, Cheng B, Miao L, Mei Y, Wu M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression.Cell Death Dis.2013;4:e574 [PubMed] https://doi.org/10.1038/cddis.2013.97
- 63. Brachmann RK. p53 mutants: the achilles’ heel of human cancers?Cell Cycle.2004;3:1030-1034. [PubMed]
- 64. Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53.Oncogene.2007;26:2202-2211. [PubMed] https://doi.org/10.1038/sj.onc.1210294
- 65. Ganci F, Sacconi A, Bossel Ben-Moshe N, Manciocco V, Sperduti I, Strigari L, Covello R, Benevolo M, Pescarmona E, Domany E, et al. Expression of TP53 mutation-associated microRNAs predicts clinical outcome in head and neck squamous cell carcinoma patients.Ann Oncol.2013;24:3082-3088. [PubMed] https://doi.org/10.1093/annonc/mdt380
- 66. Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells.Mol Cell Biol.1999;19:1438-1449. [PubMed] https://doi.org/10.1128/mcb.19.2.1438
- 67. Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L, Levine AJ, Sacchi A, Cesareni G, Oren M, Blandino G. Physical and functional interaction between p53 mutants and different isoforms of p73.J Biol Chem.2000;275:29503-29512. [PubMed] https://doi.org/10.1074/jbc.M003360200
- 68. Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M, Blandino G. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities.J Biol Chem.2002;277:18817-18826. [PubMed] https://doi.org/10.1074/jbc.M201405200
- 69. Strano S and Blandino G. p73-mediated chemosensitivity: a preferential target of oncogenic mutant p53.Cell Cycle.2003;2:348-349. [PubMed]
- 70. Guida E, Bisso A, Fenollar-Ferrer C, Napoli M, Anselmi C, Girardini JE, Carloni P, Del Sal G. Peptide aptamers targeting mutant p53 induce apoptosis in tumor cells.Cancer Res.2008;68:6550-6558. [PubMed] https://doi.org/10.1158/0008-5472.CAN-08-0137
- 71. Di Agostino S, Cortese G, Monti O, Dell’Orso S, Sacchi A, Eisenstein M, Citro G, Strano S, Blandino G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs.Cell Cycle.2008;7:3440-3447. [PubMed] https://doi.org/10.4161/cc.7.21.6995
- 72. Blagosklonny MV. p53 from complexity to simplicity: mutant p53 stabilization, gain-of-function, and dominant-negative effect.FASEB J.2000;14:1901-1907. [PubMed] https://doi.org/10.1096/fj.99-1078rev
- 73. Blagosklonny MV. Loss of function and p53 protein stabilization.Oncogene.1997;15:1889-1893. [PubMed] https://doi.org/10.1038/sj.onc.1201374
- 74. Blagosklonny MV, Giannakakou P, Romanova LY, Ryan KM, Vousden KH, Fojo T. Inhibition of HIF-1- and wild-type p53-stimulated transcription by codon Arg175 p53 mutants with selective loss of functions.Carcinogenesis.2001;22:861-867. [PubMed] https://doi.org/10.1093/carcin/22.6.861